[ad_1]
A new study has found genetic changes in a group of similar variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in India.
The researchers write:
“We believe that our report is one of the few studies that aim to provide a full picture of the evolutionary dynamics and co-mutational patterns of the strains of SARS-CoV-2 prevalent in the Indian population throughout. the year 2020. “
The study “Genomic surveillance and phylodynamic analyzes reveal the emergence of a new pattern of mutation and co-mutation in the SARS-CoV2 variants prevalent in India” is available for pre-printing on the bioRxiv* server, while the article is subject to peer review.
How they did it
Researchers carried out genomic surveillance of 54 samples positive for SARS-CoV-2 from Kolkata – the capital of West Bengal state in India. Patients ranged from 6 to 88 years with an overall male to female ratio of 1.7.
Samples were collected from August 2020 to October 2020 (Term3) and include all sequences of the SARS-CoV-2 genome in West Bengal except one.
The team notes that the genetic sequences of Term3 are different from those of India because they show a greater amount of clade “O” and clade 20A.
The Term3 sequences also differed from the genetic sequences of SARS-CoV-2 collected from April 2020 to July 2020 (Term 2). In Term2, the West Bengal region had an 82% higher amount of “G” and an 83% higher amount of clade 20A distribution than other states in India. They indicate that West Bengal viral clades evolve slowly.
Using the GSAID database, they analyzed their samples based on genomic sequences from other continents. And with the exception of North America and Europe, all other continents had prevalence of the GR clade in mid to late 2020. While North America did not show prevalence. GR, they showed a higher frequency of the GH clade. Europe showed more increase in the GV clade of SARS-CoV-2 in Europe at the end of the year.
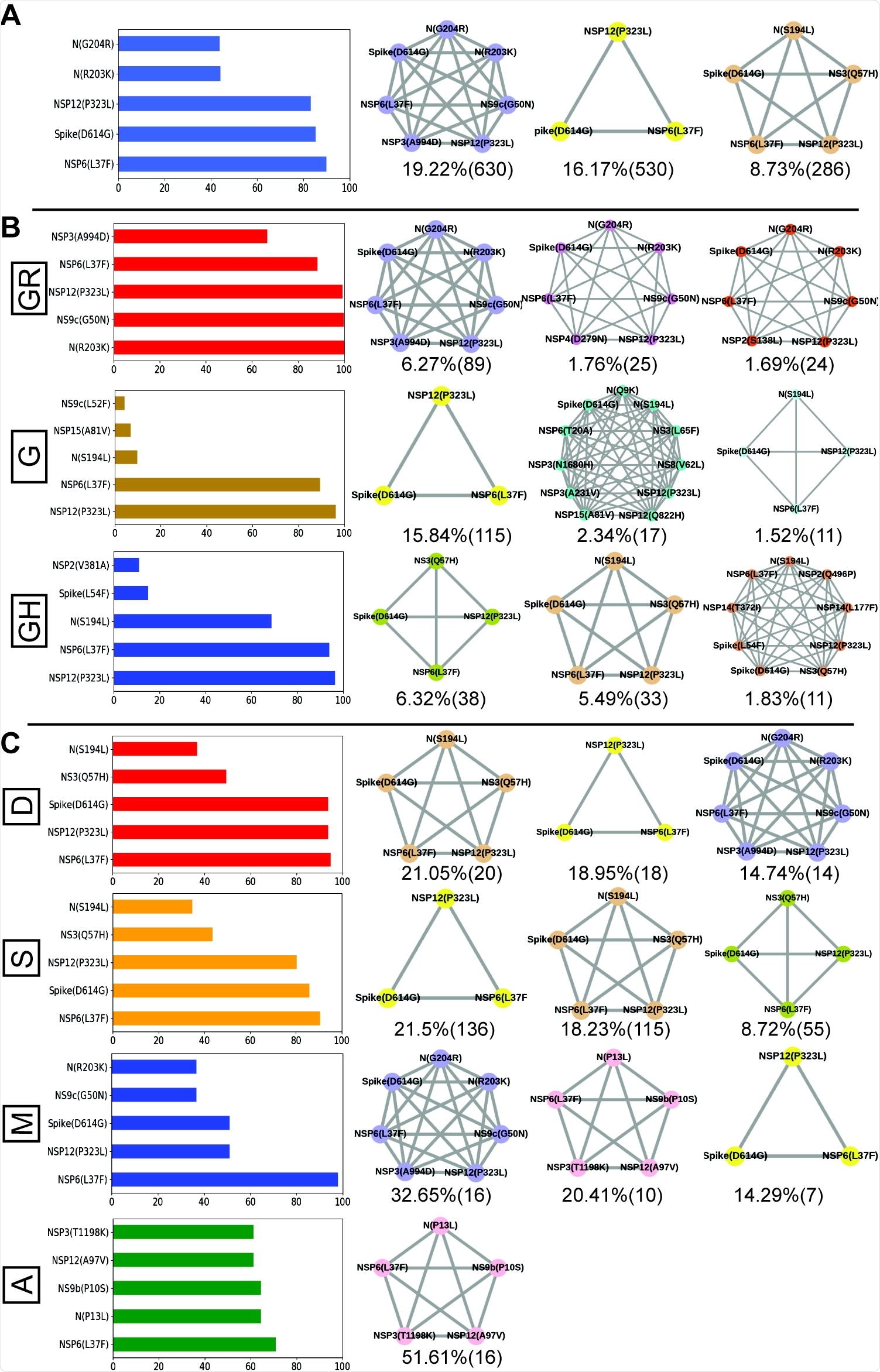
Frequent mutations, co-mutations and their association with disease severity in India. A. 5 mutations and 3 co-mutation patterns most common in India. B. The 5 most frequent mutations and 3 co-mutations in the main clades of India. Mutations defining the clade are excluded from the bar graph. C. 5 mutations and 3 co-mutations most frequent in different types of patients. Different color codes are used for specific co-mutation network patterns. The patterns of the network have been sorted and classified exclusively.
Emergence of mutations during the pandemic
Next, the researchers followed the evolutionary changes of SARS-CoV-2 in other parts of India. Genomic monitoring from viral samples from Indian patients has shown that the GR and 20B clade is more prevalent in India.
By examining the variation in co-mutations between Indian states and union territories, the researchers observed the emergence of non-clade-defining mutations during the Term2 and Term3 periods. These results were comparable at the start of the pandemic (Term1). Unique combinations of these mutations were reported more frequently towards the end of 2020.
Specifically, at Term 2, states in India such as Telangana, Maharashtra, and Karnataka showed five or more co-mutations. At the same time, the West Bengal region and Gujarat had strains of SARS-CoV-2 with less than five co-mutations.
At term 3, there were more sequences from Telangana and Maharashtra with higher co-mutant residues than those from West Bengal. The increase in distinct mutations may be associated with a significant increase in coronavirus cases in Maharashtra and Karnataka compared to West Bengal and Gujarat during the “Term2” and “Term3” periods.
“Therefore, the higher number of co-mutations in these two states could be associated with higher infectivity of the strains prevalent there.”
Mutations correlated with disease severity
The team used their genomic surveillance data to assess how the mutations affected the severity of the COVID-19 disease. They could use 806 of the 3,277 sequences as this included a patient’s COVID-19 status and whether they were deceased, symptomatic, mild, and asymptomatic.
All the sequences showed the D614G and NSP12 mutations. However, people who had symptomatic COVID-19 infection or who died from COVID-19 were more likely to have more NS9b (P10S), N (P13L), NSP12 (A97V) and NSP3 (T1198K) mutations. than asymptomatic patients.
There was also a co-mutation triad in [NSP6(L37F)-NSP12(P323L)-Spike(D614G)] in patients who have died or have mild or symptomatic COVID-19 infection.
Among 5 mutations in deceased and symptomatic patients, 3 mutations were observed in mild infection, only the NSP6 (L37F) mutation appearing in asymptomatic patients.
The co-mutation network was demonstrated in 51% of patients with asymptomatic disease and 20.41% of mild infections. However, the co-mutation network was not seen in symptomatic cases and the deceased. New co-mutation networks have been noted over time in different clades and states of India.
Researchers suggest that specific mutations and unique combinations of SARS-CoV-2 mutations correlate with disease severity based on the findings.
*Important Notice
bioRxiv publishes preliminary scientific reports which are not peer reviewed and, therefore, should not be considered conclusive, guide clinical practice / health-related behaviors, or treated as established information.
Source link
