[ad_1]
LAUSANNE, Switzerland, October 30, 2018 – Many drugs are now manufactured as powdered solids. To fully understand the behavior of active ingredients once in the body, scientists must know their exact structure at the atomic level. Researchers are therefore working on the development of technologies that make it easy to identify the exact crystalline structures of microcrystalline powders.
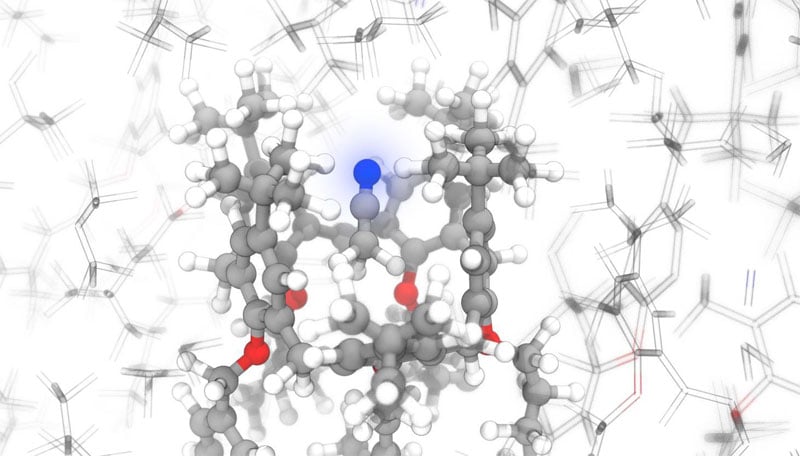
A research team from the Ecole Polytechnique Fédérale de Lausanne (EPFL) uses machine learning (ML) to rapidly predict the chemical shifts of molecular solids and their polymorphs with a precision corresponding to the theory of functional density (DFT).

Combined with experiments, the machine learning model can determine, in record time, the location of atoms in powder solids. With the kind permission of Michele Ceriotti / EPFL.
The researchers used their ML program with nuclear magnetic resonance spectroscopy (NMR) to determine the exact location of atoms in complex organic compounds. Generally, complicated and time-consuming calculations, using quantum chemistry, are needed for NMR spectroscopy to fully determine the crystal structure.
The team demonstrated that the ML model was able to determine, based on the match between the changes measured experimentally and those predicted by ML, the structures of cocaine and drugs.[4-(2-adamantylcarbamoyl)-5-tert-butylpyrazol-1-yl]benzoic acid.
According to the researchers, their approach allows computing chemical shifts for structures of ~ 100 atoms in less than a minute, thus reducing the cost of calculating chemical shift predictions in solids and reducing bottlenecks. strangulation when using chemical shifts calculated for the determination of structure in solids.
"Even for relatively simple molecules, this model is almost 10,000 times faster than existing methods and the benefit increases dramatically when one is considering more complex compounds," said Professor Michele Ceriotti. "To predict the NMR signature of a crystal of nearly 1600 atoms, our technique – ShiftML – requires about six minutes. The same feat would have taken 16 years with conventional techniques. "
The new program, which is available for free online, could benefit pharmaceutical companies, who must monitor the structure of drug molecules as active ingredients to meet patient safety requirements.
"The massive acceleration of computing times will allow us to cover much larger conformational spaces and to correctly determine structures where this was not possible before," said Professor Lyndon Emsley. "This puts most of the complex contemporary drug molecules at your fingertips."
A web version based on the protocol described here is publicly available at http://shiftml.epfl.ch. "Anyone can download a molecule and get their NMR signature in minutes," Ceriotti said.
The search was published in Nature Communications (Https://doi.org/10.1038/s41467-018-06972-x).
Source link
