
[ad_1]
The publication of drafts of the human genome in 2001 was a historic achievement1,2. Scientists were able, for the first time, to study long stretches of each human chromosome, base by base. As such, researchers were able to begin to understand how individual genes were ordered and how surrounding non-protein coding DNA was structured and organized. Despite these incredible progress, genome projects were still incomplete, with over 150 million bases missing.3. Advances in technology in the intervening years allowed researchers to complete the project, with complete sequencing of a chromosome finally being achieved.4,5 in 2020. As a result, new and uncharacterized parts of the human genome are beginning to surface, ushering in another exciting period of biological discovery.
What exactly was included in the genome project? The original project contained many previously unexplored intergenic regions. It also encompassed the vast majority of genes. The International Human Genome Sequencing Consortium1 initially estimated that the genome contained 30,000 to 40,000 genes encoding proteins, although the publication of an updated genome6 in 2004, with improved approaches to genetic prediction7, led the figure to be revised to around 20,000. The 2004 genome yielded a high resolution map of 2.85 billion nucleotides from euchromatin – the looser regions of DNA, which are enriched for genes and make up about 92% of the human genome.
The Reference Genome launched the scientific community into an era of genome exploration, shifting attention from single genes to more comprehensive genome-wide studies. However, gaps remained on each of the 23 pairs of human chromosomes, estimated to contain more than 150 megabases of unknown sequence.3 (Fig. 1). The largest gaps were in places enriched with highly repetitive DNA or sequences for which there are many nearly identical copies. These sections were originally difficult to clone, sequence and assemble properly. As a result, the Human Genome Project has intentionally under-represented these repetitive sequences. Although researchers have a very basic idea of the nature of the sequences in these regions, the high-resolution genomic organization of the regions has remained elusive.
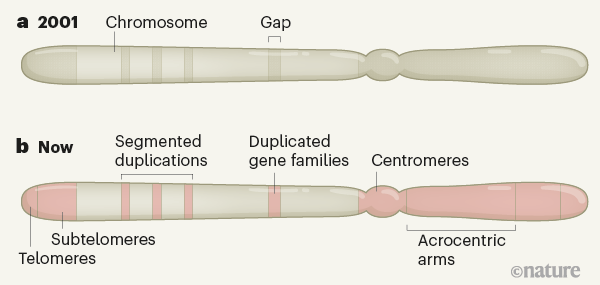
Figure 1 | Fill in the missing sequence in the human genome. a, The Human Genome Project 20011,2 covered most of the gene-rich DNA, which is loosely encapsulated in the nucleus. But many gaps remained in tightly packed regions rich in repetitive DNA sequences, which are often untranscribed (the overall extent of the gaps here is exaggerated, for ease of interpretation). bThanks to advances in sequencing and bioinformatics, researchers can now study all of these missing sequences. These include the regions of telomeres and subtelomers that cap chromosomes; centromeric structures essential for cell division; and particularly short and highly repetitive chromosomal arms called acrocentric arms. Regions in which DNA is duplicated, either in a single location or in a segmented manner, can also be analyzed.
Early attempts to fill in the gaps used long sequence reads to cover repetitive sequences – but these reads were initially very error prone. In the 2010s, new opportunities emerged, thanks to advances in the ability to read longer sequence sequences (described in references 8 and 9, for example), as well as the development of scalable bioinformatics tools. Readings of sequences of tens to hundreds of kilobases have made it possible to study the genomic organization of many medium-sized gaps. This provided information on some subtelomeric regions9 – DNA rich in repeats adjacent to the telomere structures that cap the ends of chromosomes. It also enabled the study of the first network of centromeric satellitesten, in which short sequences are repeated in tandem for about 300 kilobases. A subset of segmental duplications (sequence segments that share 90-100% of their bases and are found in multiple locations) have also been resolved, many containing genes previously absent from the reference genome.9,11. However, many of the larger regions rich in repetition of several megabase have remained insoluble.
In recent years, the combination of the two ultra-long readings9 and very precise long read data12 changed the game to solve these regions13,14, revealing, for the first time, extremely long stretches of tandem repeats and regions enriched in segmental duplications. By breaking down these technological barriers, scientists are now discovering vast regions rich in repeats that can span millions of bases and constitute the set of short arms of chromosomes.
Researchers do not yet fully understand why parts of the human genome are organized in this way. But gaining such an understanding will undoubtedly be valuable, as these repetitive-rich sequences are often placed at sites that are crucial for life. For example, long stretches of ribosomal DNA (rDNA) repeats encode RNA components of the protein synthesis machinery of the cell and play an important role in nuclear organization.15. And repeating DNA from structures called centromeres is essential for proper chromosomal segregation during cell division.16.
These large stretches of repetitive DNA come with different sets of rules, in terms of organization and genomic evolution. They are also subject to different epigenetic regulations (molecular modifications of DNA and associated proteins that do not alter the underlying DNA sequence), which causes repetitive DNA to differ from euchromatin in terms of organization, synchronization, replication and transcriptional activity.17–19. Many genome-wide tools and datasets cannot yet fully capture all of this information from highly repetitive regions of DNA, so scientists don’t yet have a complete picture of what transcription factors are. bind to them, how these regions are spatially organized in the nucleus, or how the regulation of these parts of our genome changes during development and disease states. Now, just like the initial version of the genome decades ago, researchers are faced with a new, unexplored functional landscape in the human genome. Having access to this information will spur technology and innovation to include these repeat regions, once again expanding our understanding of genome biology.
Over the past year, scientists have used extremely long and highly precise sequence reads to reconstruct entire human chromosomes from telomere to telomere.4,5. The last year also saw the publication of a nearly complete reference human genome from an effectively ‘haploid’ human cell line, with only five gaps remaining that mark the sites of the rDNA arrays (go.nature. com / 3rgz93y). In this lineage, cells have two identical pairs of chromosomes, which simplifies the challenge of repeated assembly compared to typical human cells (which are diploid, with different chromosomes inherited from mother and father). These maps together provide the first high-resolution look at centromeric regions, segmental duplications, subtelomeric repeats, and each of the five acrocentric chromosomes, which have very short arms made up almost entirely of highly repetitive DNA at one end.
It’s tempting to think that scientists are finally approaching the finish line. However, a single genome assembly, even if it is complete with near perfect sequence accuracy, is an insufficient reference to study the sequence variation that exists in the human population. Existing maps that illustrate diversity across euchromatic parts of the genome need to be extended to fully capture repeating regions, where copy number and repeat organization vary from individual to individual. To do this, it will be necessary to develop strategies for the production and routine analysis of complete human diploid genomes. The ambitious goal of achieving a fuller and more comprehensive reference of humanity will undoubtedly improve our understanding of the structure of the genome and its role in human disease, and align with the promise and legacy of the human genome project.
[ad_2]
Source link