
[ad_1]
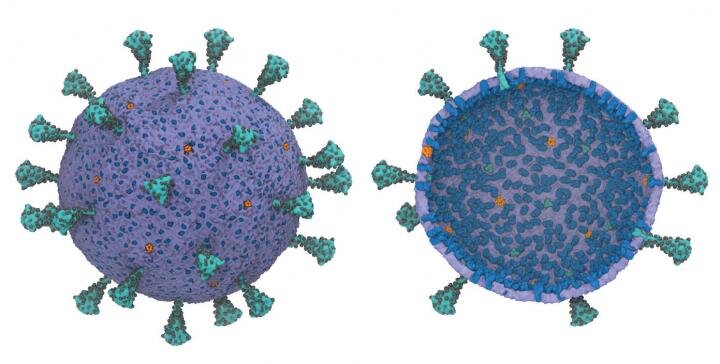
 and inner (R) views show peak protein trimers (teal), glycosylation sites (black), membrane proteins (blue), and pentameric envelope ion channels (orange). Credit: Gregory Voth, University of Chicago.")
A multiscale model of the complete SARS-CoV-2 virion has been developed for the first time using supercomputers. The model offers scientists the potential for new ways to exploit the vulnerabilities of the virus. The outer (L) and inner (R) views show the peak protein trimers (teal), glycosylation sites (black), membrane proteins (blue), and pentameric envelope ion channels (orange). Credit: Gregory Voth, University of Chicago.
The COVID-19 virus hides some mysteries. Scientists remain in the dark about aspects of how it fuses and enters the host cell; how it is assembled; and how it comes out of the host cell.
Computer modeling combined with experimental data provides information on these behaviors. But modeling over significant timescales of the SARS-CoV-2 virus that caused the pandemic has so far been limited to its components, like the spike protein, a target for the current vaccine series.
A new, coarse-grained multiscale model of the complete SARS-CoV-2 virion, its basic genetic material and its virion shell, has been developed for the first time using supercomputers. The model offers scientists the potential for new ways to exploit vulnerabilities in the virus.
“We wanted to understand how SARS-CoV-2 works holistically as a whole particle,” said Gregory Voth, Haig P. Papazian Distinguished Professor at the University of Chicago. Voth is the corresponding author of the study that developed the first comprehensive virus model, published in November 2020 in the Biophysical Journal.
“We developed a coarse-grained bottom-up model,” said Voth, “where we took information from atomistic-level molecular dynamics simulations and experiments.” He explained that a coarse-grained model only solves groups of atoms, as opposed to simulations of all atoms, where every atomic interaction is solved. “If you do it right, which is always a challenge, you keep the physics in the model.”
The first results of the study show how the spike proteins on the surface of the virus move cooperatively.
“They don’t move independently like a bunch of random, uncorrelated movements,” Voth said. “They work together.”
This cooperative movement of advanced proteins is informative of how the coronavirus explores and detects the ACE2 receptors of a potential host cell.
“The paper we published shows the beginnings of the correlation between advanced protein movement modes,” Voth said. He added that the tips are coupled to each other. When one protein moves, another moves in response as well.
“The ultimate goal of the model would be, as a first step, to study the initial attractions of virions and the interactions with ACE2 receptors on cells and to understand the origins of this attraction and how these proteins work together to go through the process of virus fusion., “Voth says.
Voth and his group have been developing coarse-grained modeling methods on viruses such as HIV and influenza for over 20 years. They “magnify” the data to make it simpler and more computationally-friendly, while remaining faithful to the dynamics of the system.
“The advantage of the coarse-grained model is that it can be computationally hundreds to thousands of times more efficient than the all-atom model,” Voth explained. The computational savings allowed the team to build a much larger model of the coronavirus than ever before, at longer timescales than was done with the all-atom models.
“What you have left are the much slower collective movements. The effects of the higher frequency, all-atom movements are built into these interactions if you do it right. That’s the idea of the systematic coarse grain.”
The holistic model developed by Voth began with atomic models of the four major structural elements of the SARS-CoV-2 virion: the tip, membrane, core and envelope proteins. These atomic models were then simulated and simplified to generate the complete grain path model.
Molecular dynamics simulations of all atoms of the peak protein component of the virion system, approximately 1.7 million atoms, were generated by study co-author Rommie Amaro, professor of chemistry and biochemistry at the University of California at San Diego.
“Their model basically ingests our data, and it can learn from the data we have at these more detailed scales and then go beyond our goal,” Amaro said. “This method developed by Voth will allow us and others to simulate over the longest timescales necessary to actually simulate the virus infecting a cell.”
Amaro elaborated on the behavior observed from coarse-grained simulations of advanced proteins.
“What he saw very clearly was the start of the S1 subunit dissociation from the tip. The entire top of the tip peels off during fusion,” Amaro said.
One of the first steps in viral fusion with the host cell is this dissociation, where it binds to the ACE2 receptor in the host cell.
“The biggest S1 opening motions they saw with this coarse-grained model were something we hadn’t yet seen in the molecular dynamics of all atoms, and in fact, it would be very difficult for us. to see, ”Amaro said. “It is an essential part of the function of this protein and of the infection process with the host cell. It was an interesting finding.”
Voth and his team used the dynamic information from all atoms about the open and closed states of the spike protein generated by the Amaro lab on the Frontera supercomputer, along with other data. The Frontera system funded by the National Science Foundation (NSF) is operated by the Texas Advanced Computing Center (TACC) at the University of Texas at Austin.
“Frontera has shown how important it is for these studies on the virus, on several scales. It was essential at the atomic level to understand the underlying dynamics of the peak with all of its atoms. There is still a lot to learn there. But now that information can be used a second time to develop new methods that allow us to come out longer and farther, like the coarse grain method, ”Amaro said.
“Frontera has been particularly helpful in providing the molecular dynamics data at the atomistic level to fuel this model. It’s very valuable,” said Voth.
The Voth group initially used the Midway2 computing cluster at the Research Computing Center at the University of Chicago to develop the coarse-grained model.
The simulations of all atoms of the membrane and envelope proteins were generated on the Anton 2 system. Operated by the Pittsburgh Supercomputing Center (PSC) with support from the National Institutes of Health, Anton 2 is a specialized supercomputer for simulations of molecular dynamics developed and provided free of charge by DE Shaw Research.
“Frontera and Anton 2 provided the main molecular-level input data into this model,” said Voth.
“One really fantastic thing about Frontera and these types of methods is that we can give people much more precise views of how these viruses move and do their jobs,” Amaro said.
“Some parts of the virus are invisible even to experience,” she continued. “And through these types of methods that we use on Frontera, we can give scientists the first important views of what these systems really look like with all their complexity and how they interact with antibodies or drugs or with parts of it. the host. cell.”
The type of information Frontera provides to researchers helps understand the basic mechanisms of viral infection. It is also helpful in designing safer and better drugs to treat and prevent disease, Amaro added.
According to Voth: “One thing that concerns us right now are the variants of SARS-CoV-2 in the UK and South Africa. Presumably, with a computing platform like the one we’ve developed here, we can quickly assess these discrepancies, which are changes. We can hopefully understand fairly quickly the changes that these mutations cause in the virus and then hopefully help design new modified vaccines in the future. ”
Scientists create first computer model of whole virus responsible for COVID-19
Alvin Yu et al, A coarse-grained multiscale model of the SARS-CoV-2 virion, Biophysical Journal (2020). DOI: 10.1016 / j.bpj.2020.10.048
Provided by the University of Texas at Austin
Quote: First full coronavirus model shows cooperation (2021, February 26) retrieved February 26, 2021 from https://phys.org/news/2021-02-coronavirus-cooperation.html
This document is subject to copyright. Other than fair use for private study or research purposes, no part may be reproduced without written permission. The content is provided for information only.
[ad_2]
Source link