
[ad_1]
With the development of DNA sequencing technology, it has become trivial to obtain the sequence of bases that encode a protein and translate it into the sequence of amino acids that make up the protein. But from there, we often find ourselves stuck. The actual function of the protein is only indirectly specified by its sequence. Instead, the sequence dictates how the chain of amino acids bends and flexes in three-dimensional space, forming a specific structure. This structure is usually what dictates the function of the protein, but obtaining it can take years of lab work.
For decades, researchers have tried to develop software that can take an amino acid sequence and accurately predict the structure it will form. Although it is a question of chemistry and thermodynamics, we had only limited success, until last year. It was at this point that Google’s DeepMind AI group announced the existence of AlphaFold, which can usually predict structures with a high degree of accuracy.
At the time, DeepMind said he would give everyone the details of his breakthrough in a future peer-reviewed article, which he finally published yesterday. Meanwhile, some university researchers grew weary of the wait, took some of DeepMind’s ideas and made their own. The document describing this effort was also released yesterday.
Dirt on AlphaFold
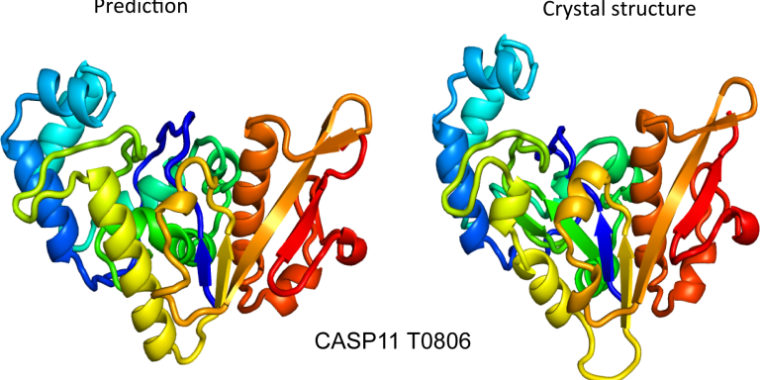
DeepMind has already described the basic structure of AlphaFold, but the new document provides much more detail. The structure of AlphaFold involves two different algorithms that communicate with each other regarding their analyzes, allowing each to refine its output.
One of these algorithms looks for sequences of proteins that are evolutionary relatives of the offending one, and it determines how their sequences line up, adjusting for small changes or even insertions and deletions. Even though we don’t know the structure of any of these parents, they can still provide significant constraints, telling us things like certain parts of the protein are still charged.
The AlphaFold team says this part of things needs around 30 related proteins to function effectively. It usually offers baseline alignment quickly and then refines it. These types of refinements may involve moving the gaps in order to place the key amino acids in the right place.
The second algorithm, which runs in parallel, splits the sequence into smaller chunks and attempts to solve the sequence of each of them while making sure that the structure of each chunk is compatible with the larger structure. This is why the alignment of the protein and its relatives is essential; if the key amino acids are in the wrong chunk, then getting the right structure will be a real challenge. Thus, the two algorithms communicate, allowing the proposed structures to return to alignment.
Structural prediction is a more difficult process, and the original ideas of the algorithm often undergo larger changes before the algorithm sets out to refine the final structure.
Perhaps the most interesting new detail in the article is where DeepMind goes by and turns off different parts of the scanning algorithms. These show that, of the nine different functions they define, all of them seem to contribute at least a little to the final precision, and only one has a dramatic effect on it. This consists in identifying the points of a proposed structure which are likely to require changes and to point them out for further attention.
The competition
In an announcement scheduled for the publication of the article, DeepMind CEO Demis Hassabis said, “We are committed to sharing our methods and providing broad and free access to the scientific community. Today, we are taking the first step towards realizing that commitment by sharing source code and publishing the full methodology of the system. “
But Google had already described the basic structure of the system, which led some researchers in academia to wonder if they could adapt their existing tools to a more structured system like DeepMind’s. And, with a seven-month lag, researchers have had ample time to act on this idea.
The researchers used DeepMind’s initial description to identify five characteristics of AlphaFold that they believed differed from most existing methods. So they tried to implement different combinations of these features and determine which ones resulted in improvements over current methods.
The easiest way to get to work was to have two parallel algorithms: one dedicated to sequence alignment, the other making structural predictions. But the team ended up dividing the structural part of things into two separate functions. One of these functions simply estimates the two-dimensional distance between different parts of the protein, and the other manages the actual location in three-dimensional space. All three exchange information, each providing guidance to the others on aspects of their task that might require further refinement.
The problem with adding a third pipeline is that it dramatically increases the hardware requirements, and academics in general don’t have access to the same kinds of IT assets as DeepMind. So while the system, called RoseTTAFold, didn’t perform as well as AlphaFold in terms of the accuracy of its predictions, it was better than any previous systems the team could test. But, given the hardware it was run on, it was also relatively fast, taking around 10 minutes when run on a 400 amino acid protein.
Like AlphaFold, RoseTTAFold breaks the protein into smaller pieces and resolves them individually before trying to assemble them into a complete structure. In this case, the research team realized that it could have an additional application. Many proteins form extensive interactions with other proteins in order to function – hemoglobin, for example, exists as a complex of four proteins. If the system is functioning as it should, feeding it two different proteins should allow it to understand their two structures. and where they interact with each other. Tests have shown that it actually works.
Healthy competition
These two articles appear to describe positive developments. For starters, the DeepMind team deserves all credit for the knowledge they had on structuring their system in the first place. Clearly, the establishment of parallel processes that communicate with each other has produced a major leap in our ability to estimate protein structures. The university team, rather than just trying to duplicate what DeepMind has done, simply adopted some of the main ideas and took them in new directions.
Right now, the two systems clearly show differences in performance, both in terms of the accuracy of their final output and in terms of the time and computational resources that must be spent on it. But with both teams seemingly engaged in the opening, there’s a good chance that the best characteristics of each could be adopted by the other.
Whatever the outcome, we are clearly in a new place from where we were just a few years ago. People have been trying to solve predictions of protein structure for decades, and our inability to do so has become more of a problem at a time when genomes provide us with large amounts of protein sequences that we are unsure of how to interpret. The demand for time on these systems is likely to be intense, as a very large part of the biomedical research community is expected to benefit from the software.
Science, 2021. DOI: 10.1126 / science.abj8754
Nature, 2021. DOI: 10.1038 / s41586-021-03819-2 (About DOIs).
[ad_2]
Source link