
[ad_1]
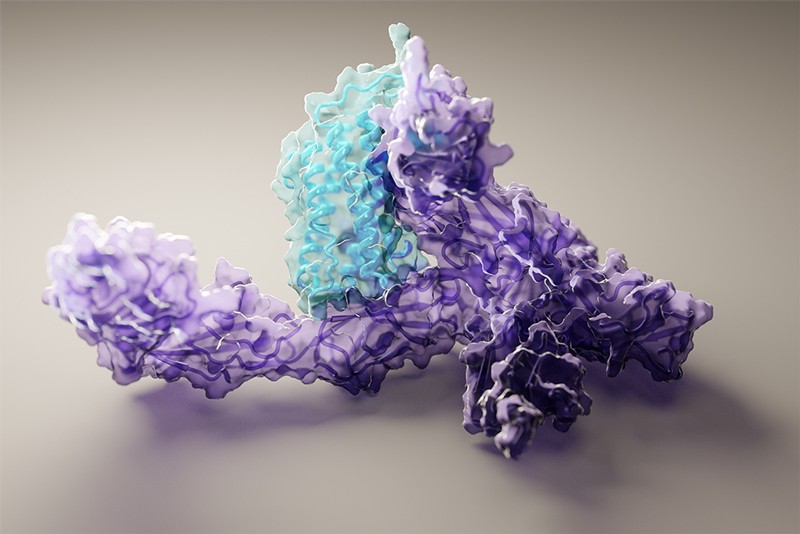
The structure of human interleukin-12 protein bound to its receptor, as predicted by machine learning software.Credit: Ian Haydon, UW Medicine Institute for Protein Design
It is the prediction of the structure of proteins for people. Software that accurately determines the 3D shape of proteins is becoming widely available to scientists.
On July 15, London-based DeepMind released an open source version of its deep learning neural network AlphaFold 2 and described its approach in an article in Nature1. The network dominated a protein structure prediction competition last year.
At the same time, an academic team developed its own protein prediction tool inspired by AlphaFold 2, which is already gaining popularity with scientists. This system, called RoseTTaFold, works almost as well as AlphaFold 2, and is described in an article in Science article also published on July 152.
The open-source nature of the tools means that the scientific community should be able to use advancements to create even more powerful and useful software, says Jinbo Xu, a computer biologist at the University of Chicago in Ill., Who did not been involved in either the effort.
Structure to function
Proteins are made up of chains of amino acids which, when folded into 3D shapes, determine the function of these proteins in cells. For decades, researchers have used experimental techniques such as X-ray crystallography and electron cryomicroscopy to determine the structures of proteins. But such methods can be time consuming and expensive, and some proteins do not lend themselves to such analysis.
DeepMind sent shockwaves into the scientific world last year when it showed that its software could accurately predict the structure of many proteins using only the sequence of proteins (which is determined by DNA). Researchers have been working on this challenge for decades, and AlphaFold 2 has performed so well in a biennial protein prediction exercise called CASP that the competition’s co-founder said “in a sense the problem is solved.”
DeepMind – who has a reputation for being suspicious of his work – described AlphaFold 2 in a brief presentation to CASP on December 1. He promised to publish an article describing the network in more detail and to make the software available to researchers, but said little else.
“Among academics, there was a fair amount of pessimism,” says David Baker, a biochemist at the University of Washington in Seattle whose team developed RoseTTaFold. “If someone has solved the problem you are working on but doesn’t reveal how they did it, how do you continue to work on it? “
“I felt like I had lost my job at the time,” explains computer chemist Minkyung Baek, a member of Baker’s team. But DeepMind’s presentation also sparked some new ideas that Baek was eager to explore. So she, Baker and their colleagues began to think about ways to replicate the success of AlphaFold 2.
They identified several key advancements, including how the network uses information about proteins related to the evolution of targets that researchers are trying to predict, and how the predicted structures of part of a protein can influence how whose network manages the sequences corresponding to other parts of the molecule.
RoseTTaFold not only performed almost as well as AlphaFold 2, but also far better than other CASP entries (including some from Baker Lab). It’s not yet clear why it couldn’t match AlphaFold 2, but DeepMind’s expertise is a possibility, Baek says. “We don’t have deep learning engineers in our lab. Xu is impressed with the efforts of Baek, Baker and their collaborators, and suspects that DeepMind’s success is due to its access to engineering expertise and superior computing power.
Fast structures
DeepMind has also streamlined AlphaFold 2. While the network used to take days of compute time to generate structures for some entries in CASP, the open source version is about 16 times faster, says AlphaFold lead researcher John Jumper. It can generate structures in minutes to hours, depending on the size of the protein. This is comparable to the speed of the RoseTTaFold.
Although the source code for AlphaFold 2 is freely available, including for commercial entities, it may not yet be particularly useful for researchers without technical expertise. DeepMind has worked with selected researchers and organizations, including the Geneva, Switzerland-based nonprofit Drugs for Neglected Diseases initiative to predict specific targets, but it hopes to expand access, said Pushmeet Kohli, Head of AI for Science at DeepMind. “There is a lot more we want to do in this space. “
In addition to making the RoseTTaFold code available for free, Baker’s team set up a server on which researchers can plug a protein sequence and obtain a predicted structure. Since launching last month, the server has predicted the structure of more than 5,000 proteins submitted by about 500 people, Baker explains.
With the code now available for free for RoseTTaFold and AlphaFold 2, researchers will be able to build on both breakthroughs, Xu says, and perhaps make the techniques fit for protein structures that AlphaFold 2 has so far struggled with. to predict. Two areas of intense interest are the prediction of the structure of complexes of multiple interacting proteins and the application of software to the design of new proteins.
[ad_2]
Source link